The world of AI-powered drug discovery keeps expanding as the capabilities of machine learning grow. One approach that seemed unthinkable just a few years ago is simulating the complicated interplays of two interlocking molecules — but that’s exactly what drug designers need to know about, and exactly what Charm Therapeutics aims to do with its DragonFold platform.
Proteins do just about everything worth doing in your body, and are the most frequent targets for drugs. And in order to create an effect, you must first understand that target, specifically how the chain of amino acids making up the protein “folds” under different circumstances.
In the recent past this was often done with complex, time-consuming X-ray crystallography, but it has recently been shown that machine learning models like AlphaFold and RoseTTAFold are capable of producing results just as good but in seconds rather than weeks or months.
The next challenge is that even if we know how a protein folds in its most common conditions, we don’t know how it might interact with other proteins let alone novel molecules made specifically to bind with them. When a protein meets a compatible binder or ligand, it can transform completely, since small changes can cascade and reconfigure its entire structure — in life this leads to things like a protein opening a passage into a cell or exposing a new surface that activates other proteins, and so on.
“That’s really where we have innovated: we have built DragonFold, which is the first protein-ligand co-folding algorithm,” said Laskh Aithani, CEO and co-founder of Charm Therapeutics.
“Designing drugs that bind to the disease-causing protein of interest very tightly and selectively (i.e., avoid binding to other similar proteins that are required for normal human functioning) is of paramount importance,” he explained. “This is done most easily when one knows how exactly these drugs bind to the protein (the exact 3D shape of the ligand bound to the disease-causing protein). This allows one to make precision modifications to the ligand such that it can bind more tightly and more selectively.”
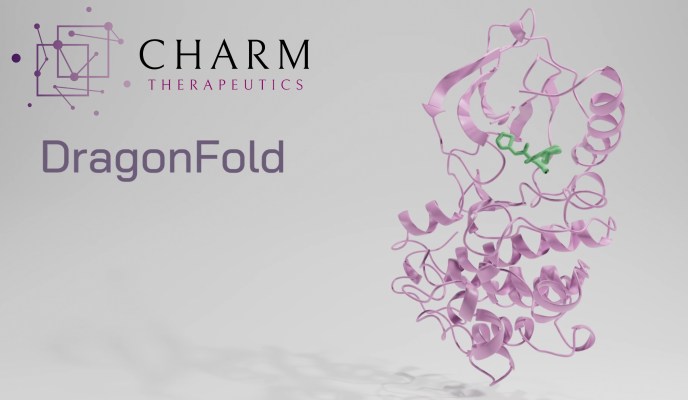
You can see a representation of this situation at the top of the article: The small green molecule and the purple protein fit together in a very specific way that is not necessarily intuitive or easy to predict. Effective and efficient simulation of this process helps screen billions of molecules, similar to earlier processes that identified drug candidates but going further and reducing the need to experimentally check whether they interact as expected.
To accomplish this, Aithani tapped David Baker, designer of the RoseTTAFold algorithm among many others and head of an influential lab at the University of Washington, to be his co-founder. Baker is well known in academia and industry as one of the leading researchers in this area, and he has published numerous papers on the subject.

Charm Therapeutics co-founders Laskh Aithani (left) and David Baker. Image Credits: Charm Therapeutics
Shortly after it was shown that algorithms could predict protein structures based on their sequence, Baker established they could also “hallucinate” new proteins that acted as expected in vitro. He’s very clearly on the leading edge here. And he won a $3 million Breakthrough prize in 2020 — definitely up to being a technical co-founder. Aithani also proudly noted the presence of DeepMind veteran Sergey Bartunov as director of AI and former pharma research lead Sarah Skerratt as head of drug discovery.
The $50 million A round was led by F-Prime Capital and OrbiMed, with participation from General Catalyst, Khosla Ventures, Braavos, Axial, and Grep VC. While such large amounts are not uncommon for software startups, it should be noted that Charm is not stopping at building the capability of characterizing these protein-ligand interactions.
The company’s early-stage funding was used to build the model, but now they’re moving on to the next step: positive identification of effective medications.
“We have the initial version [of the model] ready, and that has been validated in-silico,” Aithani said. “Over the coming quarters, we are validating it experimentally. Note that the ‘product’ will mainly be for internal use to help our own scientists discover potential medicines that we own 100% of the rights to.”
Ordinarily the testing process involves wet-lab screening of thousands upon thousands of candidate molecules, but if it works as advertised, DragonFold should massively cut down on that number. That means a relatively small lab with a relatively small budget can conceivably home in on a drug that a few years ago might require a major pharma company investing hundreds of millions.
Considering the profit profile of a novel drug, it’s no surprise that the company has attracted this kind of investment: a few tens of millions is a drop in the bucket compared with the R&D budget of any big biotech research company. All it takes is one hit and they’re laughing. It still takes a while, but AI drug discover shortens timelines as well — so expect to hear about their first candidates sooner rather than later.